
LUMASIRAN 94,5 mg-0,5 mL, solution injectable, flacon de ½ mL
Retiré du marché le : 10/03/2021
Dernière révision : 23/11/2020
Taux de TVA : 0%
Laboratoire exploitant : ALNYLAM FRANCE
Source :

LUMASIRAN est indiqué dans le traitement de l'hyperoxalurie primitive de type 1 (HP1) dans tous les groupes d'âge.
Hypersensibilité grave à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Insuffisance rénale sévère ou terminale
Le traitement par lumasiran augmente les taux plasmatiques de glycolate, ce qui peut augmenter le risque d'acidose métabolique ou aggraver une acidose métabolique pré-existante chez les patients avec une insuffisance rénale sévère ou terminale. Les signes et symptômes d'acidose métabolique doivent par conséquent être surveillés chez ces patients.
Insuffisance hépatique modérée ou sévère
Chez les patients présentant une insuffisance hépatique modérée ou sévère, il existe un risque de diminution de l'efficacité. Par conséquent, l'efficacité doit être surveillée chez ces patients (voir rubrique Propriétés pharmacocinétiques).
Excipients (sodium)
Ce médicament contient moins de 1 mmol de sodium (23 mg) par mL, c'est-à-dire qu'il est pratiquement « sans sodium ».
Résumé du profil de sécurité
L'effet indésirable le plus fréquemment rapporté (32 %) était une réaction au site d'injection.
Liste des effets indésirables
Les effets indésirables associés au lumasiran rapportés dans le cadre des études cliniques sont présentés dans le tableau ci-dessous.
Les effets indésirables sont codés en termes préférentiels (PT) dans la classe de systèmes d'organes (SOC) MedDRA et sont présentés par fréquence. La fréquence des effets indésirables est exprimée selon les catégories suivantes :
· Très fréquent (≥1/10)
· Fréquent (≥1/100 to <1/10)
· Peu fréquent (≥1/1 000 to <1/100)
· Rare (≥1/10 000 to <1/1 000)
· Très rare (<1/10 000)
Tableau 2 : Effets indésirables
| Classe de systèmes d'organes | Effet indésirable | Fréquence |
| Affections gastro-intestinales | Douleur abdominalea | Très fréquent |
| Troubles généraux et anomalies au site d'administration | Réaction au site d'injectionb | Très fréquent |
a Inclut douleur abdominale, douleur abdominale supérieure, douleur abdominale inférieure, gêne abdominale et sensibilité abdominale
b Inclut réaction au site d'injection, érythème au site d'injection, douleur au site d'injection, prurit au site d'injection, gonflement au site d'injection, gêne au site d'injection, altération de la couleur au site d'injection, masse au site d'injection, induration au site d'injection, rash au site d'injection, ecchymoses au site d'injection, hématomes au site d'injection et exfoliation au site d'injection.
Description d'effets indésirables particuliers
Réactions au site d'injection
Dans les études cliniques contrôlées contre placebo et menées en ouvert, des réactions au site d'injection ont été rapportées chez 26 patients sur 81 (32,1 %), et sont survenues pour 10 % des injections. Les symptômes les plus fréquemment rapportés étaient un érythème, une douleur, un prurit et un gonflement. La majorité des réactions au site d'injection a débuté le jour de l'administration, 7 patients ayant développé des réactions au site d'injection au moins 5 jours après l'administration (survenues dans 1,6 % des injections). Les réactions au site d'injection étaient d'intensité légère, transitoires et n'ont pas entraîné l'arrêt du traitement.
Douleurs abdominales
Dans l'étude contrôlée contre placebo, des douleurs abdominales ont été rapportées chez 1 patient sur 13 (7,7 %) sous placebo et 4 patients sur 26 (15,4 %) sous lumasiran. Dans les études contrôlées contre placebo et menées en ouvert, 17 patients sur 81 (21,0 %) ont rapporté des douleurs abdominales, dont des douleurs abdominales supérieures ou inférieures, une gêne abdominale ou une sensibilité abdominale. La plupart des cas étaient bénins, passagers et réverbles sans traitement. Aucun n'a nécessité l'interruption du traitement.
Immunogénicité
Parmi les patients atteints d'une HP1 et les volontaires sains ayant reçu LUMASIRAN, 6 personnes sur 100 (6,0 %) ont été testées positives aux anticorps anti-médicament (AAM). Les titres d'AAM étaient faibles et généralement transitoires, sans aucun impact sur l'efficacité, la sécurité d'emploi ou le profil pharmacocinétique ou pharmacodynamique du médicament.
Population pédiatrique
Le profil de sécurité du lumasiran était similaire chez les patients pédiatriques (âgés de 4 mois à 17 ans) et adultes atteints d'une HP1.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté au moyen de la fiche correspondante (voir Annexe B) de ce protocole d'utilisation thérapeutique.
Grossesse
Il n'existe pas de données sur l'utilisation du lumasiran chez la femme enceinte. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la reproduction (voir rubrique Données de sécurité précliniques). L'utilisation de ce médicament peut être envisagée pendant la grossesse en prenant en compte le bénéfice attendu pour la santé de la femme et les risques potentiels pour le foetus.
Allaitement
Il n'est pas connu si le lumasiran est excrété dans le lait maternel. Un risque pour les nouveau- nés/nourrissons ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre/de s'abstenir du traitement par lumasiran en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Il n'existe pas de données concernant les effets du lumasiran sur la fertilité chez l'Homme. Aucune incidence sur la fertilité masculine ou féminine n'a été détectée dans les études effectuées chez l'animal (voir rubrique Données de sécurité précliniques).
Aucune étude clinique d'interactions médicamenteuses n'a été réalisée (voir rubrique 5.2).
Utilisation concomitante avec la pyridoxine
L'utilisation concomitante de pyridoxine n'a pas eu d'impact significatif sur la pharmacodynamie et la pharmacocinétique du lumasiran.
Le traitement doit être instauré et supervisé par un médecin expérimenté dans la prise en charge de l'hyperoxalurie.
Posologie
LUMASIRAN est administré par injection sous-cutanée. La dose recommandée de LUMASIRAN consiste en des doses d'attaque suivies de doses d'entretien comme indiqué dans le Tableau 1. La posologie est calculée d'après le poids corporel.
Tableau 1 : Schéma posologique de LUMASIRAN en fonction du poids
| Poids corporel | Dose d'attaque | Dose d'entretien (A commencer 1 mois après la dernière dose d'attaque) |
| Moins de 10 kg | 6 mg/kg une fois par mois pendant 3 mois | 3 mg/kg une fois par mois |
| De 10 kg à moins de 20 kg | 6 mg/kg une fois par mois pendant 3 mois | 6 mg/kg une fois tous les 3 mois (posologie trimestrielle) |
| 20 kg et plus | 3 mg/kg une fois par mois pendant 3 mois | 3 mg/kg une fois tous les 3 mois (posologie trimestrielle) |
Calcul de la dose à administrer :
Poids corporel du patient (kg) x dose (mg/kg) = quantité totale (mg) du produit à administrer Calcul du volume à administrer :
Quantité totale (mg) du produit à administrer divisée par la concentration du flacon (189 mg/mL) = volume total de produit (mL) à administrer
Dose oubliée
En cas de retard ou d'oubli d'une dose, le traitement doit être administré dès que possible. L'administration doit être reprise à intervalles mensuels ou trimestriels, en fonction de la posologie prescrite, à partir de la dernière dose administrée.
Populations particulières
Patients âgés
Aucun ajustement posologique n'est nécessaire chez les patients âgés de plus de 65 ans (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
LUMASIRAN n'a pas été étudié chez les patients présentant une insuffisance hépatique. Aucun ajustement de dose n'est nécessaire chez les patients avec une élévation transitoire de la bilirubine totale (bilirubine totale > 1,0 à 1,5 fois la limite supérieure de la normale (LSN)). Une attention particulière doit être portée lors du traitement des patients présentant une insuffisance hépatique modérée ou sévère. (voir rubriques Mises en garde et précautions d'emploi et Propriétés pharmacocinétiques).
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère (débit de filtration glomérulaire estimé [DFGe] de 60 à <90 mL/min/1,73 m2) ou modérée (DFGe de 30 à <60 mL/min/1,73 m2) (voir rubrique Propriétés pharmacocinétiques). Les données cliniques disponibles sont limitées chez les patients présentant une insuffisance rénale sévère (DFGe de 15 à <30 mL/min/1,73m2), une insuffisance rénale terminale (DFGe <15 mL/min/1,73m2) ou chez les patients sous dialyse. Une surveillance est nécessaire lors du traitement chez les patients présentant une insuffisance rénale sévère ou terminale (voir rubriques Mises en garde et précautions d'emploi et Propriétés pharmacocinétiques).
Population pédiatrique
Les données sont limitées chez les patients âgés de moins d'un an. Ces patients doivent être traités avec précaution (voir rubrique Propriétés pharmacocinétiques).
Mode d'administration
Voie sous-cutanée uniquement.
Ce médicament se présente sous forme de solution prête à l'emploi dans un flacon à usage unique.
· Le volume de LUMASIRAN nécessaire doit être calculé d'après la dose recommandée en fonction du poids, comme indiqué dans le Tableau 1.
· Si la dose est supérieure à 0,5 mL (94,5 mg), plusieurs flacons seront nécessaires.
· Le volume maximal qui peut être injecté en une seule fois est de 1,5 mL. Les doses nécessitant des volumes supérieurs à 1,5 mL doivent être injectées en plusieurs fois (la dose totale doit être divisée de façon égale entre les seringues, chaque injection devant contenir à peu près le même volume) pour minimiser la gêne potentielle au niveau du site d'injection due au volume d'injection.
· Éviter de faire sortir le médicament à la pointe de l'aiguille avant que l'aiguille ne soit dans l'espace sous-cutané.
· Ce médicament doit être injecté par voie sous-cutanée dans l'abdomen, le haut du bras ou la cuisse.
· Pour les injections suivantes, il est recommandé de changer de site d'injection.
· Ce médicament ne doit pas être administré dans un tissu cicatriciel ou des zones rouges, enflammées ou gonflées.
LUMASIRAN doit être administré par un professionnel de santé. Pour les instructions relatives au produit avant administration, voir rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Durée de conservation :
3 ans.
Une fois le flacon ouvert, le médicament doit être utilisé immédiatement.
Précautions particulières de conservation :
À conserver à une température ne dépassant pas 30°C.
Conserver le flacon dans l'emballage extérieur, à l'abri de la lumière.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
En cas de surdosage, il est recommandé de suivre le patient afin de déceler tout signe ou symptôme d'effets indésirables et de lui administrer un traitement symptomatique approprié.
Classe pharmacothérapeutique : non encore attribuée, code ATC : non encore attribué. Mécanisme d'action
Le lumasiran est un petit acide ribonucléique interférent (pARNi) à double brin, qui réduit les taux de l'enzyme glycolate-oxydase (GO) en ciblant l'acide ribonucléique messager (ARNm) du gène de l'hydroxyacide oxydase 1 (HAO1) dans les hépatocytes par interférence ARN. La diminution des taux de l'enzyme GO réduit la quantité disponible de glyoxylate, un substrat pour la production d'oxalate. Ce phénomène conduit à une réduction des taux urinaires et plasmatiques d'oxalate, la cause sous- jacente des manifestations de la maladie chez les patients atteints d'une HP1. Comme l'enzyme GO est en amont de l'enzyme déficiente alanine-glyoxylate aminotransférase (AGT) responsable de l'HP1, le mécanisme d'action de Lumasiran est indépendant de la mutation du gène AGXT sous-jacente.
Efficacité clinique
L'efficacité du lumasiran a été démontrée dans une étude clinique randomisée, en double aveugle et contrôlée contre placebo, conduite chez des patients âgés de 6 ans et plus et atteints d'une HP1 (ILLUMINATE-A) ainsi que dans une étude clinique monobras conduite chez des patients âgés de moins de 6 ans et atteints d'une HP1 (ILLUMINATE-B).
ILLUMINATE-A
Au total, 39 patients atteints d'une HP1 ont été randomisés selon un rapport de 2/1 pour recevoir des doses sous-cutanées de lumasiran ou de placebo pendant la période de 6 mois en double aveugle contrôlée contre placebo. Les patients de 6 ans et plus ayant un DFGe ≥30 mL/min/1,73 m2 ont été recrutés et ont reçu 3 doses d'attaque de 3 mg/kg de lumasiran ou un placebo administré une fois par mois, suivies de doses d'entretien trimestrielles de 3 mg/kg de lumasiran ou de placebo (voir rubrique Posologie et mode d'administration). À l'issue de la période de 6 mois de traitement en double aveugle, les patients, y compris ceux affectés initialement au placebo, sont entrés dans une période d'extension au cours de laquelle ils ont reçu le lumasiran.
Pendant la période de 6 mois en double aveugle contrôlée contre placebo, 26 patients ont reçu le lumasiran et 13 ont reçu le placebo. L'âge médian des patients lors de l'administration de la première dose était de 14,9 ans (min-max : 6,1 - 61,0 ans), 66,7 % des patients étaient de sexe masculin et 76,9 % des patients étaient d'origine caucasienne. La valeur médiane de l'oxalurie des 24 heures corrigée en fonction de la surface corporelle (SC) à l'inclusion était de 1,72 mmol/24 h/1,73 m2, le rapport médian oxalate /créatinine sur un échantillon d'urine à l'inclusion était de 0,21 mmol/mmol, et la valeur médiane du taux plasmatique d'oxalate à l'inclusion était de 13,1 µmol/L. Au total, 33,3% des patients avaient une fonction rénale normale (DFGe ≥90 mL/min/1,73 m2), 48,7% avaient une insuffisance rénale légère (DFGe de 60 à <90 mL/min/1,73 m2), et 18% avaient une insuffisance rénale modérée (DFGe de 30 à <60 mL/min/1,73 m2). Parmi les patents inclus dans l'étude, 84,6% avaient des antécédents d'événements de calculs rénaux symptomatiques et 53,8% avaient des antécédents de néphrocalcinose, au moment de l'inclusion. Les bras de traitement étaient équilibrés à l'inclusion en termes d'âge, de taux urinaire d'oxalate et de DFGe.
Le critère d'évaluation principal était le pourcentage de réduction par rapport à l'inclusion de l'oxalurie des 24 heures corrigée en fonction de la SC, en moyenne sur les Mois 3 à 6. Le lumasiran a été associé à une réduction statistiquement significative de 65,4 % de l'oxalurie des 24 heures corrigée en fonction de la SC versus 11,8% dans le groupe placebo représentant une différence de 53,5% (IC 95 % : 44,8 ; 62,3 ; p <0,0001). Comme pour le critère d'évaluation principal, une réduction à 6 mois de 60,5% du rapport oxalate/créatinine sur un échantillon d'urine, a été observée dans le groupe lumasiran, comparée à une augmentation de 8,5% dans le groupe placebo. En outre, les patients traités par lumasiran ont présenté une diminution rapide et durable de l'oxalurie des 24 heures corrigée en fonction de la SC, comme indiqué sur la Figure 1.
Figure 1 : ILLUMINATE-A : Pourcentage de variation par rapport à l'inclusion de l'oxalurie des 24 heures corrigée en fonction de la SC par mois
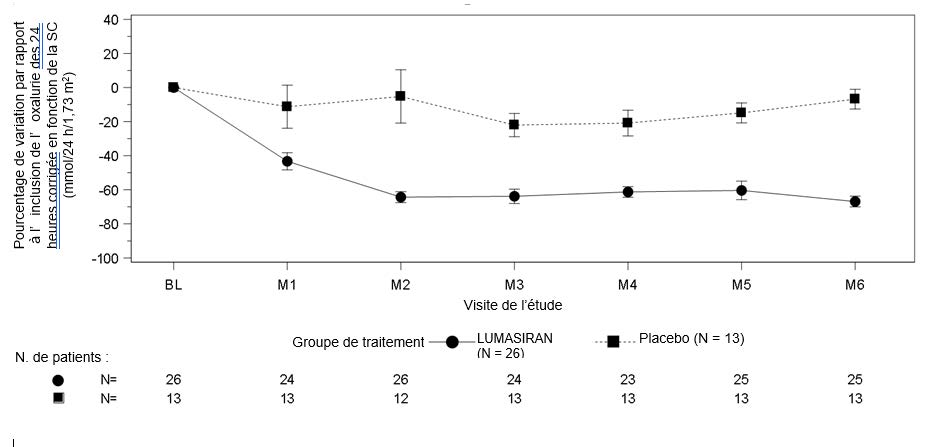
Abréviations : BL = Inclusion ; M = mois ; ETM = erreur type de la moyenne, SC = Surface Corporelle. Les résultats sont exprimés en moyenne (± ETM) de pourcentage de variation par rapport à l'inclusion.
Au Mois 6, une plus grande proportion de patients traités par lumasiran que de patients sous placebo ont atteint des niveaux normaux ou quasi normaux d'oxalurie des 24 heures corrigée en fonction de la SC (≤1,5 × LSN), comme indiqué dans le Tableau 3.
Tableau 3 : ILLUMINATE-A Résultats des critères d'évaluation secondaires sur la période de 6 mois en double aveugle contrôlée contre placebo
| Critères d'évaluation | Lumasiran (N = 26) | Placebo (N = 13) | Différence entre les traitements (IC 95 %) | Valeur de p |
| Proportion de patients présentant une oxalurie des 24 h inférieure ou égale à la LSN‡ | 0,5 (0,3 ; 0,7)§ | 0 (0 ; 0,2)§ | 0,5 (0,2 ; 0,7)Ꝕ | 0,001# |
| Proportion de patients présentant une oxalurie des 24 h inférieure ou égale à 1,5 × LSN‡ | 0,8 (0,6 ; 1,0)§ | 0 (0 ; 0,2)§ | 0,8 (0,5 ; 0,9)Ꝕ | <0,0001# |
| Moyenne du pourcentage de réduction de l'oxalémie par rapport à l'inclusion*Þ | 39,8 (2,9)† | 0,3 (4,3)† | 39,5 (28,9 ; 50,1) | <0,0001 |
Abréviations : LSN = limite supérieure de la normale ; ETM = erreur type de la moyenne
Les résultats sont basés sur une analyse par chromatographie en phase liquide couplée à une spectrométrie de masse en tandem (LC-MS/MS)
* Estimation basée sur la moyenne, sur les Mois 3, 4, 5 et 6, de la moyenne des moindres carrés du pourcentage de réduction, à l'aide d'un modèle mixte pour mesures répétées.
† Moyenne des MC (ETM).
‡ LSN = 0,514 mmol/24 h/1,73 m2 pour l'oxalurie des 24 heures corrigée en fonction de la SC.
§ IC 95 % basé sur l'intervalle de confiance exact de Clopper-Pearson.
ꝔCalcul utilisant la méthode Newcombe basée sur la méthode de score de Wilson
# La valeur de p est basée sur un test de Cochran-Mantel-Haenszel stratifié en fonction de la valeur à l'inclusion de l'oxalurie des 24 heures corrigée en fonction de la SC (≤1,70 vs >1,70 mmol/24 h/1,73 m2).
Þ Analysé chez 23 patients sous Lumasiran et 10 patients sous placebo, dont la valeur à l'inclusion permettait d'envisager une réduction
La réduction par rapport à l'inclusion de l'oxalurie des 24 heures corrigée en fonction de la SC observée chez les patients atteints d'une HP1 et recevant le lumasiran versus placebo était similaire dans tous les sous-groupes prédéfinis selon l'âge, le sexe, l'origine ethnique, , la présence d'une insuffisance rénale, l'utilisation de pyridoxine (Vitamine B6) à l'inclusion, et les antécédents d'événements de calculs rénaux symptomatiques (Figure 2).
Figure 2 : ILLUMINATE-A : Pourcentage de variation par rapport à l'inclusion de l'oxalurie des 24 heures corrigée en fonction de la SC, analyse en sous-groupes
| Sous-groupe | Lumasiran - Placebo | Lumasiran | Placebo | |
| | | (N) | (N) | |
| Ensemble | 26 | 13 | ||
| Âge à la sélection | | | ||
| 6 - < 12 ans | 9 | 7 | ||
| 12 - < 18 ans | 5 | 1 | ||
| ≥ 18 ans | 12 | 5 | ||
| Sexe | | | ||
| Masculin | 18 | 8 | ||
| Féminin | 8 | 5 | ||
| Origine | | | ||
| Caucasienne | 21 | 9 | ||
| Non caucasienne | 5 | 4 | ||
| Prise de pyridoxine à l'inclusion | | | ||
| Oui | 13 | 9 | ||
| Non | 13 | 4 | ||
| Oxalate urinaire sur 24 heures corrigé en fonction de la SC à l'inclusion | | | ||
| ≤ 1,70 mmol/24 h/1,73 m² | 11 | 7 | ||
| > 1,70 mmol/24 h/1,73 m² | 15 | 6 | ||
| DFGe à l'inclusion | | | ||
| < 60 mL/min/1,73 m² | 4 | 3 | ||
| ≥ 60 mL/min/1,73 m² | 22 | 10 | ||
| Antécédents de calculs rénaux symptomatiques | | | ||
| Oui | 23 | 10 | ||
| Non | 3 | 3 | ||
| | En faveur du lumasiran | En faveur du placebo | | |
Les taux réduits d'oxalate observés dans la période de double aveugle ont été maintenus jusqu'à 12 mois, durant la période d'extension de l'étude.
Le DFGe et les événements de calculs rénaux (signalés en événements par 100 personnes-jours) ont été évalués durant les périodes en double aveugle et en extension pour un total de 12 mois. Le DFGe est resté stable chez les patients sous lumasiran. Dans le groupe lumasiran, le taux d'événements de calculs rénaux signalés 12 mois avant le consentement était de 0,87 (IC 95 % : 0,70 ; 1,08). Les événements observés durant la période de double aveugle et les 6 premiers mois de la période d'extension étaient respectivement de 0,30 (IC 95 % : 0,17 ; 0,51) et 0,23 (IC 95 % : 0,13 ; 0,43). Dans le groupe placebo, le taux d'événements de calculs rénaux signalés 12 mois avant le consentement était de 0,15 (IC 95 % : 0,07 ; 0,31) et les événements observés durant la période de double-aveugle étaient 0,18 (IC 95 % : 0,07 ; 0,48). Durant les 6 premiers mois de traitement par lumasiran lors de la période d'extension, 0,05 (IC 95 % : 0,01 ; 0,32) événements ont été observés chez les patients traités précédemment par le placebo. Concernant la néhrocalcinose, les données jusqu'au 6è mois de la période en double aveugle sont disponibles. Sur les 34 patients ayant fait l'objet d'une échographie rénale à l'inclusion et au Mois 6, 3 sur 22 ont présenté une amélioration de la néphrocalcinose dans le groupe lumasiran, et 1 sur 12 ont présenté une aggravation de la néphrocalcinose dans le groupe placebo. Aucun des autres patients sous lumasiran (n=19) ou placebo (n=11) n'a présenté de modification de la néphrocalcinose.
ILLUMINATE-B
Un total de 18 patients ont été inclus et traités avec le lumasiran dans une étude multicentrique monobras en cours, conduite chez des patients atteints d'une HP1 (ILLUMINATE-B). L'étude a recruté des patients âgés de moins de 6 ans avec un DFGe >45 mL/min/1,73 m2 chez les patients âgés de 12 mois et plus, et une créatinine sérique normale chez les patients âgés de moins de 12 mois. Dans
l'analyse primaire sur 6 mois, à la première dose, 3 patients pesaient moins de 10 kg, 12 pesaient de 10 kg à moins de 20 kg, et 3 pesaient 20 kg et plus lors de l'administration de la première dose. L'âge médian des patients lors de l'administration de la première dose était de 51,4 mois (min-max : 4,0- 74,0 mois), 55,6 % étaient de sexe féminin, et 88,9 % étaient d'origine caucasienne. La valeur médiane du rapport urinaire oxalate/créatinine à l'inclusion était de 0,47 mmol/mmol.
Au Mois 6, les patients traités par le lumasiran ont atteint une réduction de 72,0% (IC 95 % : 66,4 ; 77,5) du rapport urinaire oxalate/créatinine par rapport à l'inclusion (moyenne sur les Mois 3 à 6), soit le critère d'évaluation principal. Le lumasiran était associé à une réduction rapide et prolongée du rapport urinaire oxalate/créatinine (Figure 3), de manière similaire dans tous les groupes de poids. Le pourcentage de réduction de l'excrétion de l'oxalate urinaire était cohérent par rapport aux données d'ILLUMINATE-A.
Figure 3 : ILLUMINATE-B : Pourcentage de variation par rapport à l'inclusion du rapport oxalate/créatinine sur un échantillon d'urine par mois
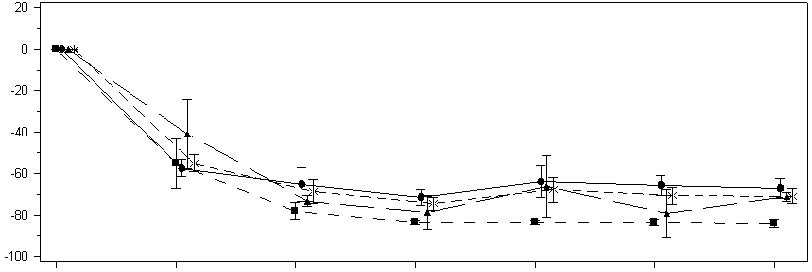
| Pourcentage d'évolution par rapport à l'inclusion du rapport urinaire oxalate/ créatinine (mmol/mmol) | | |||||||
| | | BL | M1 | M2 | M3 | M4 | M5 | M6 |
| | | | | | Visite | | | |
| | | Groupe de poids initial dans ILLUMINATE-B | - - -■- - - < 10 kg | —●— 10 à < 20 kg | - -▲- - ≥ 20 kg | -----Û Tous traités par lumasiran | ||
| Nb de patients : | ||||||||
| ■ | N = | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| ● | N = | 12 | 12 | 12 | 12 | 12 | 12 | 12 |
| ▲ | N = | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| Û | N = | 18 | 18 | 18 | 18 | 18 | 18 | 18 |
Au 6 mois, 9 patients ont atteint une quasi-normalisation (≤1.5×LSN) du rapport urinaire oxalate/créatinine, dont 1 patient qui a atteint la normalisation (≤LSN),
En outre, une réduction moyenne de l'oxalate plasmatique de 31,7% (IC 95 % : 23,9 ; 39,5) a été observé au Mois 6 par rapport à l'inclusion (moyenne des Mois 3 à 6). Durant la période de 6 mois, le DFGe est resté stable, et 2 événements de calculs rénaux post-inclusion ont été signalés, comparé aux 4 événements de calculs rénaux survenus chez 3 patients dans les 12 mois précédant le consentement. 14 patients sur 18 avaient une néphrocalcinose à l'inclusion. Les données d'échographie rénale au Mois 6 ont montré une amélioration chez 8 patients, incluant 3 patients avec une amélioration bilatérale. Aucun des 18 patients n'a eu un début ou une dégradation de néphrocalcinose.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec LUMASIRAN dans un ou plusieurs sous-groupes de la population pédiatrique dans l'hyperoxalurie (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
Après administration sous-cutanée, le lumasiran est rapidement absorbé, avec un temps médian (intervalle) nécessaire pour atteindre la concentration plasmatique maximale (tmax) de 4,0 (0,5-12,0) heures. Chez les enfants et adultes ≥20 kg avec HP1, au niveau de dose recommandé de 3 mg/kg, la concentration plasmatique maximale (Cmax) et l'aire sous la courbe des concentrations à partir du temps zéro jusqu'à la dernière concentration mesurable après l'administration (ASC0-dernière) du lumasiran étaient respectivement de 529 (205-1 130) ng/mL et 7 400 (2 890-10 700) ng·h/mL. Chez les enfants pesant moins de 20 kg, la Cmax et l'ASC0-dernière du lumasiran après la dose recommandée de lumasiran de 6 mg/kg étaient respectivement de 912 (523-1 760) et 7 960 (5 920-13 300). Les concentrations de lumasiran ont été mesurables jusqu'à 24 à 48 heures après la dose.
Distribution
Sur des échantillons de volontaires sains adultes, la liaison du lumasiran aux protéines plasmatiques est modérée à forte (77 à 85 %) aux concentrations cliniquement pertinentes. Pour un patient adulte atteint de HP1, l'estimation de population du volume central apparent de distribution (Vd/F) pour le lumasiran est de 4,9 L. La distribution du lumasiran s'effectue principalement dans le foie après administration sous-cutanée.
Biotransformation
Le lumasiran est métabolisé par les endo et exo-nucléases en oligonucléotides de plus faible longueur. Les études in vitro indiquent que le lumasiran n'est pas métabolisé par les enzymes CYP450.
Élimination
Le lumasiran est éliminé du plasma principalement par absorption hépatique, seulement 7 à 26 % de la dose administrée étant retrouvée dans les urines sous forme de lumasiran, d'après les données de volontaires sains adultes, et de patients atteints d'HP1 de > 6 ans. La demi-vie plasmatique terminale moyenne (%CV) du lumasiran est de 5,2 h (47,0 %). L'estimation de population de la clairance plasmatique apparente était de 26,5 L/h pour un adulte type de 70 kg. La clairance rénale moyenne du lumasiran était mineure et allait de 2,0 à 3,4 L/h, chez des patients pédiatriques et adultes atteints d'HP1..
Linéarité/non-linéarité
Le lumasiran a présenté une pharmacocinétique linéaire à légèrement non-linéaire, et indépendante du temps dans le plasma après des doses sous-cutanées uniques allant de 0,3 à 6 mg/kg et des doses répétées de 1 et 3 mg/kg une fois par mois ou de 3 mg/kg une fois tous les 3 mois. Aucune accumulation de lumasiran n'a été observée dans le plasma après l'administration répétée d'une dose mensuelle ou trimestrielle.
Relation(s) pharmacocinétique/pharmacodynamique
La concentration plasmatique de lumasiran ne reflète pas la durée de l'activité pharmacodynamique du lumasiran. Une absorption rapide et ciblée du lumasiran par le foie provoque une baisse rapide des concentrations plasmatiques. Dans le foie, le lumasiran possède une demi-vie longue conduisant à un maintien de l'effet pharmacodynamique pendant l'intervalle de doses mensuel ou trimestriel.
Interactions
Des études in vitro suggèrent que le lumasiran n'est pas un substrat ou un inhibiteur des enzymes du cytochrome P450 (CYP). Il n'est pas attendu que le lumasiran inhibe ou induise les cytochromes ou module l'activité des transporteurs.
Populations particulières
Patients âgés
Aucune étude n'a été conduite chez les patients âgés de ≥ 65 ans. L'âge n'était pas une co-variable significative dans la pharmacocinétique du lumasiran.
Sexe et origine ethnique
Dans les études cliniques, il n'y avait pas de différence au niveau de l'exposition plasmatique ou de la pharmacodynamie du lumasiran en fonction du sexe ou de l'origine ethnique.
Insuffisance hépatique
Aucune étude n'a été conduite chez les patients présentant une insuffisance hépatique (voir rubrique Posologie et mode d'administration). Les données pharmacocinétiques limitées chez les patients présentant une élévation légère et transitoire de la bilirubine totale (bilirubine totale >1,0 à 1,5 x LSN) ont montré une exposition plasmatique du lumasiran comparable et une pharmacodynamie similaire à celles des patients avec une fonction hépatique normale. La littérature publiée montre une expression plus faible des récepteurs des asialoglycoprotéines dans le foie, c'est-à-dire les récepteurs responsables de l'absorption du lumasiran, chez les patients atteints d'insuffisance hépatique. Des données non cliniques suggèrent que cela peut ne pas influencer l'absorption hépatique ou la pharmacodynamie aux doses thérapeutiques. La pertinence clinique de ces données est inconnue.
Insuffisance rénale
Les patients présentant une insuffisance rénale légère (DFGe de 60 à <90 mL/min/1,73 m2) avaient une exposition plasmatique au lumasiran comparable et une pharmacodynamie similaire à celles des patients présentant une fonction rénale normale (DFGe ≥90 mL/min/1,73 m2). Chez les patients avec une insuffisance rénale modérée (DFGe de 30 à <60 mL/min/1,73 m2) la Cmax était similaire à celle des patients ayant une fonction rénale normale ; l'ASC était 25 % plus élevée selon des données limitées. Des données cliniques limitées sont disponibles chez des patients avec une insuffisance rénale sévère (DFGe <30 mL/min/1,73 m2), une insuffisance rénale terminale (DFGe
< 15 mL/min/1,73 m²), ou étant sous dialyse (voir rubrique Posologie et mode d'administration). Pour les patients en insuffisance rénale terminale sous dialyse, pour la même catégorie de poids corporel, une élévation transitoire de la Cmax de 3 à 7 fois, et une augmentation de ASC0-dernière de 2 à 3,5 fois ont été observées (voir rubrique Propriétés pharmacocinétiques Relation(s) Pharmacocinétique/Pharmacodynamie). Cependant, la décroissance des taux plasmatiques sous le seuil de détection sous 24 à 48 heures était similaire par rapport aux patients sans insuffisance rénale.
Population pédiatrique
Les données chez les enfants de moins de 1 an sont limitées. Chez les enfants < 20 kg, la Cmax du lumasiran était 2 fois plus élevée, en raison de la dose nominalement supérieure de 6 mg/kg et de la vitesse d'absorption plus rapide. La pharmacodynamie du lumasiran était comparable chez les patients pédiatriques (âgées de 4 mois à 17 ans) et les patients adultes, en dépit des concentrations plasmatiques transitoires plus élevées chez les enfants < 20 kg, en raison de la distribution rapide et prédominante du lumasiran vers le foie.
Poids corporel
Le schéma posologique recommandé produisait une Cmax jusqu'à 2 fois plus élevée chez les enfants pesant < 20 kg, tandis que l'ASC était similaire pour les poids corporels étudiés (6,2 à 110 kg).
LUMASIRAN n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité et de génotoxicité n'ont pas révélé de risque particulier pour l'Homme.
Chez les rats, mais pas chez les singes, des changements microscopiques dans le foie (par ex., vacuolisation hépatocellulaire, mitose et caryomégalie) ont été observés, accompagnés d'une baisse des taux de fibrinogène plasmatique et d'autres modifications biologiques. La raison de cette spécificité apparente aux rongeurs n'est pas comprise et sa pertinence envers l'Homme reste floue.
Le lumasiran n'a produit aucun effet indésirable sur la fertilité des mâles et des femelles et sur le développement pré-natal et post-natal des rats. Des anomalies squelettiques ont été observées lors d'études sur le développement embryo-foetal de rats et de lapins, mais à des multiples d'exposition élevés par rapport aux expositions thérapeutiques humaines. Les niveaux sans effet nocif observé (NOAEL) étaient environ 20 à 70 fois plus élevées (d'après des expositions mensuelles).
Une étude de toxicité de détermination de dose réalisée sur des rats nouveau-nés n'a pas montré de sensibilité accrue chez le rat en développement du fait de la toxicologie ou de la pharmacologie du lumasiran à des multiples d'exposition de 2 par rapport aux expositions thérapeutiques humaines (d'après des expositions mensuelles).
Aucune étude n'a été effectuée chez l'animal en vue d'évaluer le potentiel carcinogène du lumasiran.
Ce médicament est prêt à l'emploi et à usage unique exclusivement. Voie sous-cutanée uniquement
• Avant l'administration, le matériel, non inclus dans la boîte, nécessaire à l'administration et qui comprend une seringue stérile (0,3 mL, 1 mL ou 3 mL), une aiguille de calibre 18 (G) et une aiguille de calibre 25 G à 31 G, doit être rassemblé.
• Le volume requis de LUMASIRAN doit être calculé d'après la dose recommandée en fonction du poids (voir rubrique Posologie et mode d'administration).
• Une aiguille de calibre 18G doit être utilisée pour prélever LUMASIRAN dans le flacon. Le flacon doit être tenu à la verticale ou légèrement incliné et le bord plat de l'aiguille doit être dirigé vers le bas.
• Pour les volumes inférieurs à 0,3 mL, une seringue stérile de 0,3 mL est recommandée.
• Le médicament doit être administré avec une aiguille stérile de calibre 25 à 31 G et de longueur 13 mm ou 16 mm pour injection sous-cutanée.
• Remarque : Ce médicament ne doit pas être poussé dans l'aiguille de calibre 25 G à 31 G.
• Les seringues, aiguilles de transfert et aiguilles d'injection ne doivent être utilisées qu'une seule fois.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament soumis à prescription hospitalière
Prescription réservée aux spécialistes en néphrologie ou en pédiatrie.
Prescription réservée aux spécialistes en néphrologie ou en pédiatrie.
Solution injectable.
Solution limpide, incolore à jaune.
Flacon en verre scellé par un bouchon en élastomère halobutyle revêtu de fluoropolymère et une capsule en aluminium à soulever. Chaque flacon contient 0,5 mL de solution injectable.
Conditionnement de 1 flacon.
Chaque mL de solution contient du lumasiran sodique équivalent à 189 mg de lumasiran. Chaque flacon de 0,5 mL contient 94,5 mg de lumasiran.
Excipient à effet notoire :
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Hydroxyde de sodium (ajustement du pH)
Acide phosphorique (ajustement du pH)
Eau pour préparations injectables
Acide phosphorique (ajustement du pH)
Eau pour préparations injectables